caused by an immaturity of the enzyme glucuronyl transferase
should resolve in 5 – 7 days
jaundice that persists for > 2 weeks is considered pathologic
Differential Diagnosis and Diagnostic Evaluation
if the unconjugated bilirubin fraction is > 80%, differential diagnosis includes Rh and ABO
incompatibilities, hemolytic disease, metabolic disorders, TORCH
if the conjugated bilirubin fraction is > 20%, then the etiology of the jaundice is
cholestasis or obstruction
bile in the duodenum or green or brown stools eliminates obstruction
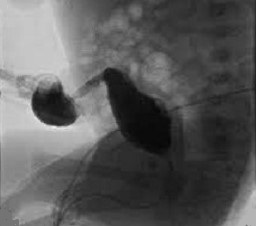
HIDA scan can rule out obstruction if the isotope appears in the intestine
Biliary Atresia
Pathogenesis
intrauterine fibrosis of both the intrahepatic and extrahepatic biliary tree
exact etiology is unclear: both infectious and immunologic mechanisms have been proposed
Clinical Manifestations
persistent and progressive jaundice
hepatomegaly is the most common physical finding
growth retardation and portal hypertension may develop in the later stages
Diagnosis
serum bilirubin and alkaline phosphatase levels will be elevated
on ultrasound, the presence of a gallbladder does not rule out biliary atresia
the intrahepatic bile ducts are never dilated in biliary atresia
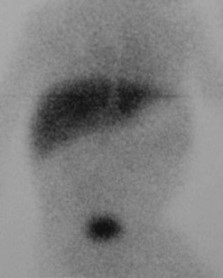
HIDA scan is an accurate way of diagnosing obstruction
percutaneous liver biopsy demonstrating proliferating bile ducts, portal fibrosis, and
cholestasis may be valuable in difficult cases
HIDA Scan showing no excretion into the biliary system
Treatment
Kasai Hepatoportoenterostomy
should be performed within the first 2 months of life to get the best results
fibrous tissue at the porta hepatis contains microscopically patent bile ductules
that communicate with the intrahepatic ductal system
Kasai’s procedure entails dividing this fibrous tissue and draining it into a
Roux-en-Y limb of jejunum
complications include cholangitis and progressive liver disease
at least 70% of patients will ultimately require a liver transplant
Liver Transplantation
primary treatment if the diagnosis is delayed until the infant is 4 months of age
or greater
salvage procedure if the infant does not improve after the Kasai procedure
majority of patients who undergo the Kasai procedure will require a liver transplant
by age 10
Choledochal Cysts
Pathogenesis
etiology is controversial
one theory is an abnormal biliary-pancreatic junction: formation of a common channel with
gradual weakening of the bile duct wall by pancreatic enzyme destruction leading to
dilatation, inflammation, and finally cyst formation
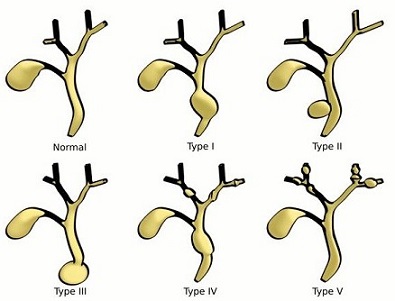
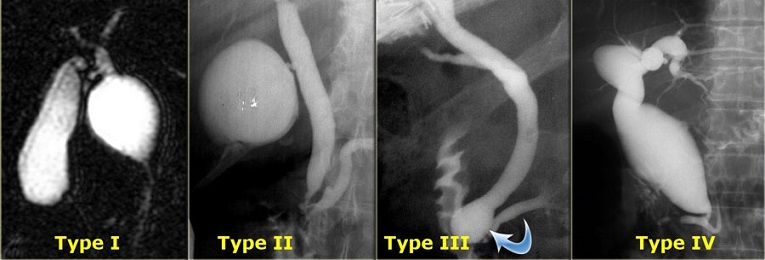
5 main variants: most common type is fusiform dilatation (type I)
other associated biliary-pancreatic anomalies are common
females > males (4:1)
Clinical Manifestation
50% of patients present with jaundice, a right upper quadrant mass, and abdominal pain
many patients present with episodic abdominal pain over months or years
if undetected, sequelae include cholangitis, cirrhosis, and portal hypertension
Diagnosis
now frequently diagnosed by prenatal US
CT or MRCP will confirm the diagnosis and demonstrate the dimensions of the cyst as well as its
relationship to the portal vein and hepatic artery
ERCP is reserved for confusing cases
Management
Cystoenterostomy
internal drainage procedure using the duodenum or jejunum
no longer recommended
serious complications include cholangitis and malignant degeneration of the cyst
wall
Resection
procedure of choice is complete resection of the cyst with a Roux-en-Y
hepaticojejunostomy
distal end of the bile duct is oversewn at the duodenum
cyst must be carefully resected from the anterior wall of the portal vein
an abnormally high insertion of the pancreatic duct must be identified to prevent
injury
if there is a lot of pericystic inflammation, the outer posterior layer of the cyst
wall can be left in place on the portal vein
Pediatric Abdominal Wall Defects
Embryology
abdominal wall is formed by four separate embryologic folds (cephalic, caudal, right and left
lateral folds) that join to form a large umbilical ring
between the 5th and 10th weeks of fetal development the intestinal tract undergoes rapid growth
outside of the abdominal cavity within the proximal umbilical cord
as development is completed, the intestine gradually returns to the abdominal cavity
contraction of the umbilical ring completes the process of abdominal wall formation
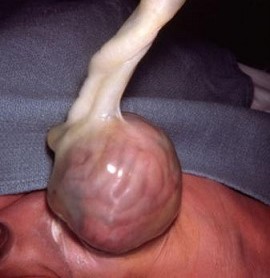
Omphalocele
Pathology
covered defect of the umbilical ring into which abdominal contents (bowel and solid viscera)
herniate
results from a failure of migration of the lateral folds
giant omphaloceles are > 4 cm in diameter and often contain liver
sac is composed of an outer layer of amnion and an inner layer of peritoneum
occurs in ~ 1 in 5000 births
high incidence of associated anomalies (cardiac, chromosomal)
Management
Emergency Management
orogastric tube should be inserted to decompress the stomach and prevent swallowed
air from causing bowel distention
sac should be covered with a sterile dressing or bowel bag
no effort should be made to reduce the sac since this may lead to rupture of the sac,
respiratory distress, or decreased venous return
IV antibiotics should be administered
must avoid hypothermia
Operative Management
Small Defects
may be closed primarily or with mesh
Large Defects
closed in stages, using a Silastic ‘silo’ as a temporary housing for
the bowel
the silo is secured to the edge of the defect with a running suture
each day the silo is gradually reduced in size in the neonatal ICU until the
defect can be formally closed
Nonoperative Management
usually reserved for patients with other life-threatening anomalies
consists of topical application of an escharotic agent (Mercurochrome, Silvadene)
allows the sac to thicken and epithelialize
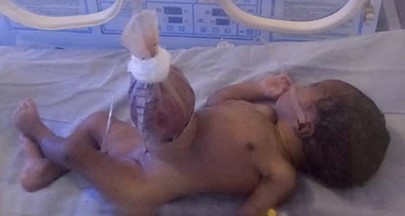
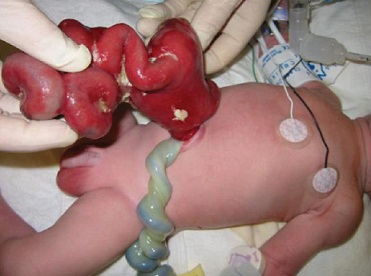
Gastroschisis
Pathology
defect of the anterior abdominal wall just to the right of the umbilicus
umbilical cord remains intact
considered to be a hernia of the umbilical cord that ruptures
no associated sac
exposed bowel is very thickened and congested
incidence of associated anomalies is relatively infrequent (except for intestinal atresia,
which occurs in 10% to 15% of cases)
Management
neonate must be fluid resuscitated as a result of third-space deficits from sequestration of
fluid in the edematous bowel
lower half of the baby (including the eviscerated bowel) is placed in a sterile bowel bag
primary closure is possible in most cases
if primary closure is not possible, then a temporary silo may be placed, and the viscera
gradually reduced
because of the low incidence of severe anomalies, the survival rate is much better than
with omphalocele
Patent Urachus
communication between the bladder and the umbilicus
first sign is moisture or urine coming from the umbilicus
diagnosis may be confirmed by cystogram
treatment is excision of the tract with closure of the bladder
Umbilical Hernia
results from failure of closure of the umbilical ring
hernias < 1 cm in size usually close spontaneously by 4 - 5 years of age
since incarceration is rare, surgical repair is reserved for large hernias or those that do not
close spontaneously by age 5
the defect is closed primarily
Inguinal Hernia
Pathogenesis
results from failure of closure of the processus vaginalis, which should close a few months
before birth
male:female ratio is 10:1
risk of incarceration is high because of the narrow inguinal ring
Diagnosis
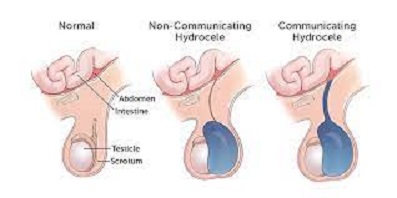
can be difficult to distinguish between an inguinal hernia and a hydrocele, even with
transillumination
Management
Incarcerated Hernia
should try to reduce the hernia by sedating the infant and applying bimanual pressure
if reduction is successful, repair can be performed within 24 hours
if reduction is unsuccessful, or if a bowel obstruction is present, then emergency
repair should be performed
Reducible Hernia
elective repair should be planned since spontaneous resolution does not occur
treatment consists of high ligation of the sac; a floor repair or ring-tightening
procedure is rarely required
the distal sac is widely opened (to prevent a hydrocele) and left in situ
(to prevent an ischemic testicle)
what to do about the opposite side, if anything, remains controversial
laparoscopic evaluation of the opposite side through the hernia sac is gaining
popularity
Hydrocele
if no hernia is present, then observation until the child reaches 12 months of age
is the proper approach
after the child reaches 12 months of age, an elective hydrocelectomy should be
performed