associated with congenital anomalies such as aniridia, Beckwith-Wiedemann syndrome,
hemihypertrophy, horseshoe kidney, urinary tract defects
specific Wilms’ tumor suppressor genes (WT-1, WT-2) located on chromosome 11p have been
discovered and cloned
Clinical Manifestations
~ 500 cases/year in the U.S.
peak age of incidence is between 1 and 5 years
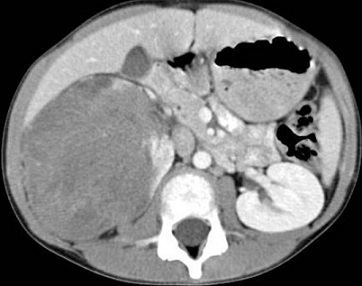
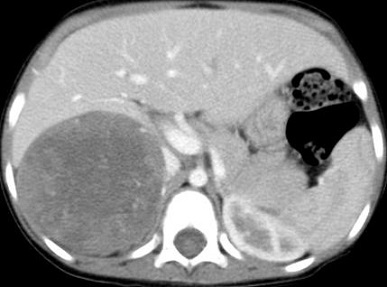
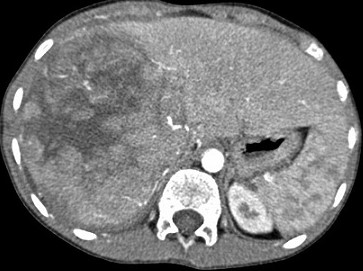
usually presents as a hard, asymptomatic abdominal or flank mass
hematuria may occur in 10% to 15% of cases
hypertension occurs in ~ 20%
13% are bilateral
Diagnostic Workup
ultrasound will demonstrate a solid intrarenal lesion and will also demonstrate whether the
mass has extended into the renal vein, IVC, or right atrium
CT scan of the abdomen should be obtained to determine the status of the other kidney as
well as to evaluate for both local invasion of contiguous structures and distant metastases
(liver, periaortic nodes)
chest x-ray and chest CT scan should be done to evaluate for pulmonary metastases
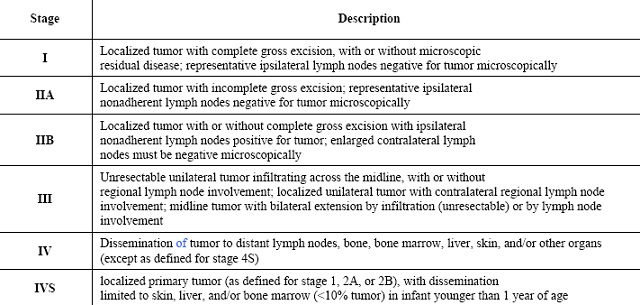
National Wilms Tumor Study Staging
Treatment
Surgery
radical nephroureterectomy is performed through a transverse abdominal incision
opposite kidney and liver are carefully evaluated for tumor involvement
whenever possible the renal hilum should be clamped before the tumor is mobilized,
both to limit blood loss and to prevent tumor embolization
if contiguous organs are involved, they should be resected en bloc if possible
perihilar and periaortic nodes are removed for staging purposes
tumor rupture/spillage results in local recurrence and must be avoided
Adjuvant Chemotherapy
depends both on the stage and histology (divided into favorable and unfavorable)
actinomycin-D and vincristine are the primary agents used
in advanced stages or in those with unfavorable histology, Adriamycin and radiation
are added to the treatment regimen
overall survival for favorable histology tumors for all stages is 90%
even in stage IV disease, cure rates of 80% are achieved
Neoadjuvant Chemotherapy
used to shrink massive tumors in order to make resection possible
in Europe, neoadjuvant chemotherapy is used in most patients
Treatment of Bilateral Wilms’ Tumors
goal is to preserve renal parenchyma and avoid bilateral nephrectomy
treatment options include: bilateral heminephrectomy, total nephrectomy on one side
and partial nephrectomy on the other side, and neoadjuvant chemotherapy
Neuroblastoma
Pathology
embryonal tumor of neural crest origin
may arise in any sympathetic ganglion
most common sites include: adrenal medulla (50%), para-aortic ganglia (24%), posterior
mediastinum (20%), neck (3%), and pelvis (3%)
may secrete a variety of hormones, including catecholamines, VIP
Clinical Manifestations
50% occur by 2 years of age and 90% by 8 years
symptoms depend on the site of the primary and the presence of metastases
abdominal tumors usually present as asymptomatic abdominal masses
thoracic tumors may present with respiratory distress or Horner’s syndrome
paraplegia or cauda equina syndrome is related to extradural extension of tumor
hypertension is present in 20% to 35% of cases
at presentation, only 50% have localized disease
Diagnostic Workup
plain x-rays, US, and CT scans are the initial studies obtained
CT scan can usually discern between a renal tumor and a neuroblastoma
24-hour urine collection for VMA, HVA, and metanephrines show elevated levels in > 85% of
cases
MRI is used to rule out spinal involvement
bone scan should be done to rule out bone metastases and a bone marrow biopsy should be done
to rule out marrow involvement
Staging
Treatment
Surgery
complete surgical excision is the best hope for cure
abdominal tumors are approached through a transverse incision
thoracic tumors are approached through a posterolateral thoracotomy
tumors with a spinal component require laminectomy and intraspinal removal
for infants with stage IVs disease, surgery is not recommended since there is a high
rate of spontaneous regression
Chemotherapy
used in a neoadjuvant role for locally unresectable tumors
used as adjuvant therapy for resected tumors with unfavorable risk factors
(> 10 copies of N-myc)
used in a therapeutic role for patients with metastatic disease
immunotherapy is now being used in high-risk patients
Outcome
age and disease stage are the 2 most important prognostic factors
N-myc amplification is associated with poor prognosis, regardless of age
overall 5-year survival rates are ~ 74%
Rhabdomyosarcoma
Pathology
arises from mesenchymal tissues
most common sites are: head and neck (36%), extremities (19%), genitourinary tract (21%),
trunk (9%)
invades local structures early
metastasis occurs via hematogenous and lymphatic spread
Clinical Manifestations
depends on the site of origin
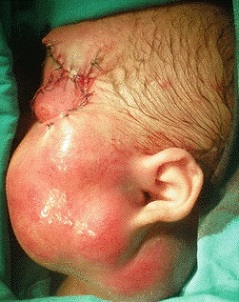
Rhabdomyosarcoma of the Head and Neck
Diagnostic Workup
CT scan of the involved area
bone marrow biopsy
tissue diagnosis may be made by an incisional or excisional biopsy
Management
wide local excision with negative margins is the optimal procedure
amputations and exenterations can often be avoided with neoadjuvant chemotherapy and radiation
Hepatic Neoplasms
Hemangioma
most common benign liver tumor in infancy
Clinical Manifestations
most present as painless abdominal masses
some infants develop cardiac failure secondary to A-V shunting within the liver
Diagnosis
CT scan with bolus IV contrast is usually diagnostic
Management
only symptomatic infants need to be treated since most lesions will spontaneously
involute
treatment includes corticosteroids, diuretics, and digoxin
alfa-interferon may cause involution
rarely, hepatic resection or hepatic artery ligation may be necessary
Malignant Tumors
Pathology
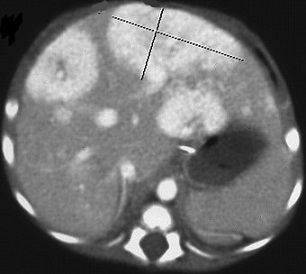
hepatoblastoma is the most common malignant tumor in children and is usually
diagnosed before 4 years of age
hepatocellular carcinoma is the next most common malignant tumor and has a peak
incidence between 10 and 15 years of age
sarcomas are rare lesions
Clinical Manifestations
usually presents as a painless abdominal mass
jaundice is not usually present
Diagnostic Workup
AFP is elevated in 90% of children with hepatoblastoma
CT scan is essential for evaluating resectability as well as multicentricity and
involvement of the contralateral lobe
US is valuable for detecting tumor extension into the hepatic veins and IVC
Hepatoblastoma
Management
complete surgical resection is the treatment of choice, usually requiring lobectomy
or trisegmentectomy
unresectable lesions may benefit from neoadjuvant chemotherapy
liver transplantation is an option for some locally unresectable tumors
Outcome
~ 70% of hepatoblastoma patients are long-term survivors
~ 25% of hepatocellular carcinoma patients are long-term survivors
Teratomas
Pathology
composed of tissue from all 3 germ layers (endoderm, ectoderm, and mesoderm)
80% occur in females
may be benign or malignant
may arise in any part of the body
usually found in midline structures
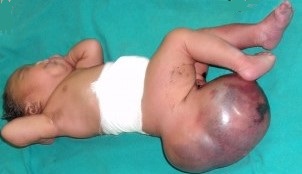
most common location is sacrococcygeal (60%)
Clinical Manifestations
thoracic teratomas present as an anterior mediastinal mass
ovarian teratomas present as an abdominal mass and may have symptoms of torsion, bleeding,
or rupture
sacrococcygeal teratomas present as a large sacral mass in the newborn period
Management of Sacrococcygeal Teratomas
most tumors are diagnosed on prenatal US or at birth
complete resection should be performed as early as possible because of the risk of malignant
degeneration
coccyx should be resected because of the risk of tumor recurrence
rectal and genital structures can usually be preserved
tumors with significant intra-abdominal extension will require a combined posterior and
intra-abdominal approach
hemorrhage is the major postoperative complication
Management of Ovarian Teratomas
accounts for 25% of childhood teratomas
25% present with torsion
definitive treatment is oophorectomy or salpingo-oophorectomy
ascites or peritoneal washings should be sent for cytology