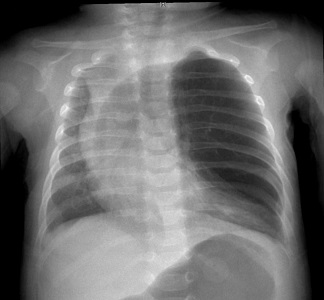
results from failure of closure of the pleuroperitoneal canal in the developing fetus
80% - 90% occur on the left side
the resulting posterolateral diaphragmatic defect is known as a Bochdalek hernia
abdominal contents herniate into the thoracic cavity, compressing the ipsilateral developing
lung and resulting in pulmonary hypoplasia
the contralateral lung is also affected
pulmonary hypertension also develops by increased thickness of arteriolar smooth muscle
morbidity and mortality of CDH is dependent on the severity of pulmonary hypoplasia and
pulmonary hypertension
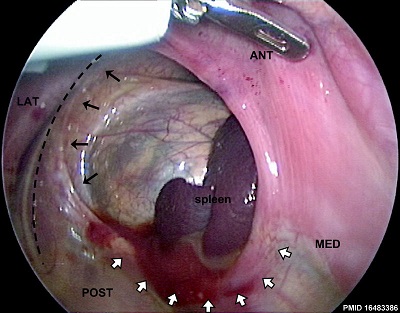
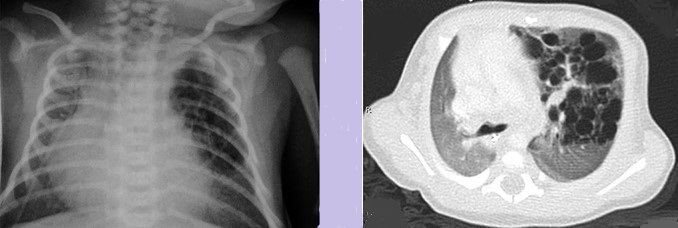
Thoracoscopic View of a Congenital Diaphragmatic Hernia
Clinical Presentation
presenting symptom at birth is respiratory distress
a scaphoid abdomen with decreased bowels sounds is a common finding
bowel sounds may be auscultated in the chest, and the heart may be shifted over to the right
persistent pulmonary hypertension results in a severe right to left shunt
Diagnosis
majority of cases are now diagnosed on prenatal ultrasound, often as early as 15 weeks
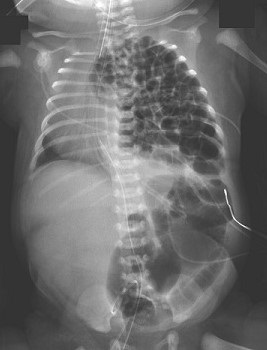
CXR shows multiple bowel loops in the chest and a mediastinal shift
Management
Antenatal Surgery
open fetal surgery has not improved survival and is no longer performed
in Europe, laparoscopic occlusion of the fetal trachea, which stimulates lung growth
by accumulation of lung fluid, is undergoing a clinical trial
Stabilization
airway must be immediately secured
high airway pressures must be avoided, since they can cause barotrauma and decreased
venous return to the heart
permissive hypercapnia is associated with less barotrauma and improved survival
an orogastric tube is placed to decompress gastric distention, which can worsen lung
compression, mediastinal shift, and the ability to ventilate
nitric oxide is used for pulmonary vasodilation
Sildenafil treats pulmonary hypertension by relaxing pulmonary artery smooth muscle
infants with profound respiratory distress may need extracorporeal membrane
oxygenation (ECMO)
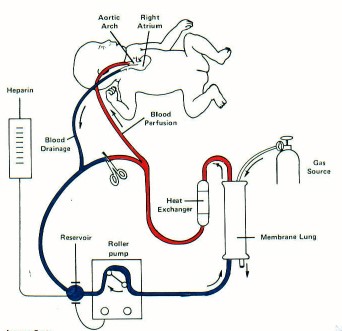
ECMO
desaturated blood is removed, oxygen and carbon dioxide are exchanged
through the membrane oxygenator, oxygenated blood is then returned to the
circulation
criteria include normal cardiac anatomy as determined by echocardiography,
absence of fatal chromosomal abnormalities, and the expectation that the
infant would die without ECMO
relative contraindications to ECMO include weight < 2 kg and gestational age
< 34 weeks
access may be by the venovenous route (right internal jugular vein) or
venoarterial (internal jugular vein, carotid artery) route
in most infants, within 7 – 10 days, pulmonary hypertension resolves, and
lung function improves enough to wean off bypass
since patients require systemic anticoagulation, bleeding complications can
be life-threatening
Surgery
Timing of Surgery
infants with relatively stable pulmonary status may undergo repair on day
2 – 4
timing of surgery for infants on ECMO is controversial, but recent reports
suggest that repair after ECMO is associated with better outcomes
Procedure
approached through a subcostal incision
the abdominal contents are returned to the abdomen
if there is loss of domain, a temporary silo or skin-only closure may be
necessary
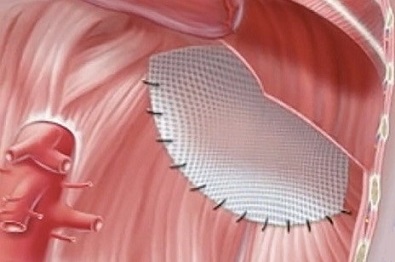
small diaphragmatic defects may be closed primarily, but larger defects will
require a prosthetic patch (usually Gore-Tex)
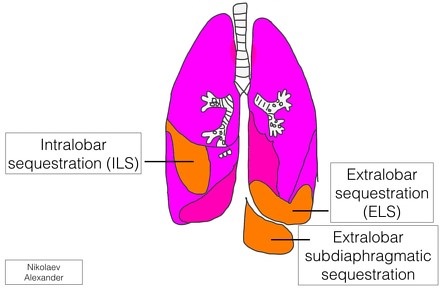
Pulmonary Sequestration
Pathogenesis
mass of lung tissue without connections to the pulmonary artery or tracheobronchial tree
blood supply originates from the aorta; venous drainage may be systemic or pulmonary
usually occurs in the left lower chest
2 kinds of sequestration: extralobar and intralobar
extralobar sequestration is nonaerated lung separated from the main lung mass and encased in
its own pleura
intralobar sequestration occurs within the parenchyma of the lung
Clinical Manifestations
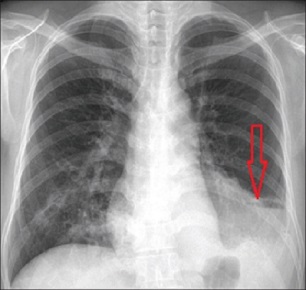
extralobar sequestration is usually asymptomatic and is discovered incidentally on chest x-ray
intralobar sequestration may cause repeated pulmonary infections
unusual complications can include infarction, hemoptysis, or left-to-right cardiac shunt
Diagnosis
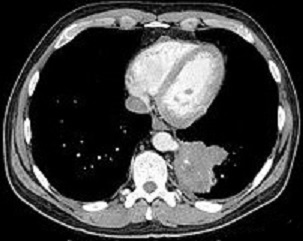
diagnosis of extralobar sequestration can be confirmed by CT scan
US with color Doppler or arteriogram will demonstrate the systemic blood supply of
intralobar sequestration
Extralobar Sequestration
Management
if the diagnosis is secure, resection of extralobar sequestration is not necessary
treatment of intralobar sequestration usually requires a left lobectomy
Bronchogenic Cyst
Pathogenesis
may occur anywhere along the respiratory tract, but are usually found near the carina and
right hilum
consists of a single cyst lined with respiratory epithelium and containing cartilage and
smooth muscle
cysts within the pulmonary parenchyma usually communicate with a bronchus; those within the
mediastinum usually do not
Clinical Manifestations
may be completely asymptomatic
in the neck, they may produce symptoms of airway compression
in the lung parenchyma, they may cause infection or obstruction with atelectasis
Diagnosis
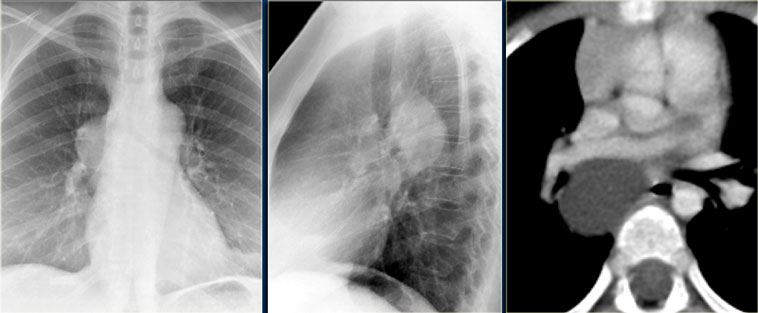
chest x-ray will show a dense cyst
CT scan will define the precise location of the mucus-filled cystic mass
Management
surgical resection is recommended, even for asymptomatic cysts
Congenital Lobar Emphysema
Pathogenesis
progressive hyperexpansion of one or more lobes of the lung, causing atelectasis of the
adjacent lobes
caused by intrinsic bronchial obstruction from poor cartilage development or extrinsic
compression
Clinical Manifestations
symptoms range from asymptomatic to mild respiratory distress to respiratory failure
Diagnosis
chest x-ray shows a hyperlucent affected lobe with adjacent lobar compression and varying
degrees of shift of the mediastinum and compression of the opposite lung
bronchoscopy is not advised because it can produce more air trapping and cause
life-threatening respiratory distress in a stable infant
Management
asymptomatic, incidentally found lesions can be observed, because some lesions will
spontaneously regress
lobectomy is required for symptomatic lesions
Congenital Pulmonary Airway Malformation (CPAM)
Pathogenesis
hamartomatous lesions in which a multicystic mass replaces normal lung tissue
blood supply is pulmonary
connected to the tracheobronchial tree
Clinical Manifestations
majority are asymptomatic in infancy, but some may present with life-threatening respiratory
distress in the perinatal period
may cause recurrent pneumonia or chronic cough
may undergo malignant transformation (rhabdomyosarcoma)
Diagnosis
may be diagnosed prenatally by U/S and follow-up MRI
in symptomatic patients, CXR reveals a cystic thoracic mass, occasionally with
air-fluid levels
CT scan is confirmatory
Management
for CPAM diagnosed prenatally, some reports suggest steroids may promote spontaneous regression
emergent resection is indicated in infants who present with acute respiratory distress
in asymptomatic infants, resection is generally performed at 6 months because of the risk of
infection and malignant transformation
Foreign Bodies
Pathogenesis
most commonly occurs in the toddler age group
peanuts and popcorn are the most common foodstuffs aspirated
most common location of the foreign body is the right main stem bronchus or the right lower
lobe
the lodged foreign body causes air trapping, leading to atelectasis and infection
Clinical manifestations
the child usually coughs or chokes while eating
if the object lodges in the trachea, complete respiratory obstruction may occur
objects that lodge more distally may initially be asymptomatic until pneumonia supervenes
a unilateral wheeze may be heard audibly or on auscultation
Diagnosis and Management
nuts, seeds, plastic toy parts are radiopaque on CXR
CXR may show hyperexpansion of the affected lobe on expiration
rigid bronchoscopy confirms the diagnosis and allows for retrieval of the object
Esophageal Disorders
Esophageal Atresia and Tracheoesophageal Fistula
Pathology
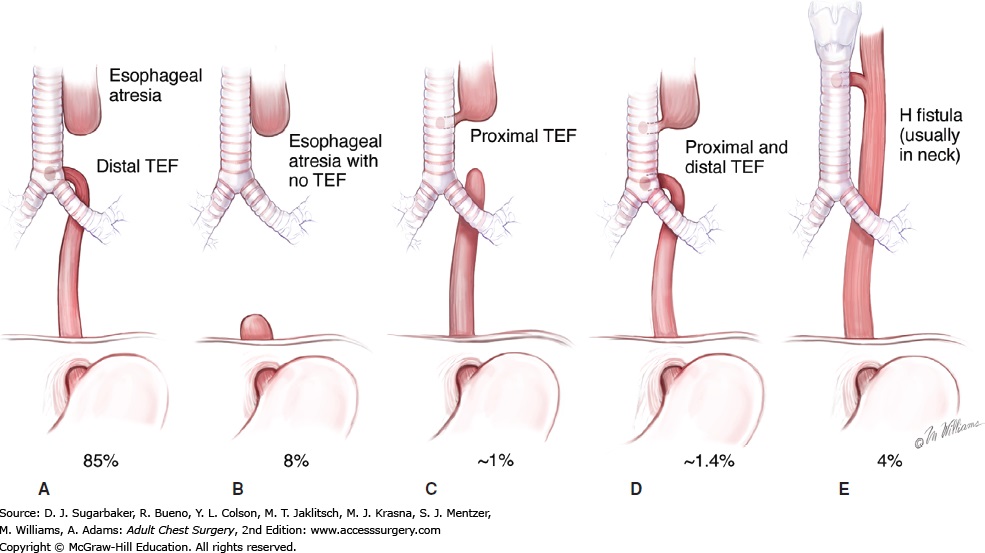
5 recognized anatomic variants
10% have the VATER syndrome (vertebral, anal, TE fistula, radial limb or renal anomalies)
20% have congenital heart disease
maternal history of polyhydramnios is common
Clinical manifestations
earliest signs are regurgitation, excessive drooling, and coughing or choking at first feeding
abdominal distention is often prominent because air passes from the fistula into the stomach
respiratory distress, aspiration, and pneumonia may occur as gastric juice refluxes from the
stomach into the lungs through the fistula
Diagnosis
esophageal atresia is strongly suggested by the inability to pass a N-G tube into the stomach
contrast studies are discouraged because of the risk of aspiration
if gas is present in the GI tract, an associated TE fistula must also be present
bronchoscopy localizes the level of the fistula and excludes upper pouch fistulas
echocardiography should be done to exclude cardiac defects
Initial Management
NG tube on suction in the upper pouch
head up prone position to minimize reflux and aspiration
infant warmer
establish IV access
IV antibiotics (even if pneumonia is not present)
avoid mechanical ventilation, if possible, because positive pressure ventilation will direct
air through the fistula preferentially
Surgical Management
stable infants undergo immediate surgical repair
unstable infants undergo delayed repair
Primary Repair of Proximal Atresia with Distal Fistula
performed through a right extrapleural thoracotomy
azygous vein must be divided to expose the fistula
fistula is transected and the defect in the trachea closed and buttressed with
mediastinal pleura
upper pouch is dissected superiorly into the neck to obtain adequate length for the
anastomosis
if there is too large a gap between the two ends, a proximal circular myotomy is
performed to gain additional length
end-to-end anastomosis is performed in one or two layers
Delayed Repair
reserved for infants with serious coexisting anomalies, extreme prematurity, or
respiratory distress
proximal pouch is kept on suction
gastrostomy tube is placed for decompression
rarely, ligation of the fistula is required to stabilize the pulmonary status
esophageal anastomosis is carried out once the infant has been stabilized
Repair of Isolated Esophageal Atresia
diagnosed by a blind proximal pouch and an absence of air below the diaphragm
typically there is a long gap between the proximal and distal pouches
most surgeons perform a gastrostomy for feedings and wait 6 to 10 weeks for the upper
pouch to elongate
daily dilatations of the upper pouch may help to gain additional length
at the time of thoracotomy, myotomies of both the upper and lower pouches are often
necessary to make the anastomosis possible
if it is impossible to bring the two ends together, an esophagostomy is made out of
the upper pouch and esophageal replacement will have to be performed at 1 year of
age with a colon interposition
Repair of H-Type Fistula
both the trachea and esophagus are normal
fistula usually can be divided through a neck incision
placing strap muscle between the esophageal and tracheal suture lines promotes
healing and prevents recurrence of the fistula
Complications
mortality rate is directly related to associated cardiac defects and chromosomal
abnormalities
if no other anomalies are present, survival is > 95%
Anastomotic Leak
occurs in 10% to 20% of cases
since the repair is done by the extrapleural approach, leak does not result
in empyema
as long as the anastomosis is adequately drained, spontaneous closure is the
rule
Anastomotic Stricture
occurs in 15% to 30% of cases
endoscopic dilatation usually is sufficient to manage the problem
Gastroesophageal Reflux
occurs in 25% to 50% of cases
Nissen fundoplication is often necessary
since esophageal motility is poor in patients with esophageal atresia, an
antireflux procedure can result in esophageal obstruction
Gastroesophageal Reflux (GER)
Clinical Presentation
symptoms vary, depending upon the age of the patient and underlying medical conditions
vomiting is a common complaint
growth retardation from calories loss is one of the most serious complications
recurrent pneumonia, bronchitis, and asthma symptoms may be associated with chronic GER
also may contribute to near-miss sudden infant death syndrome
Diagnosis
a detailed history will strongly suggest GER
contrast esophagraphy will provide anatomic and functional data (motility, gastric emptying)
24-hour esophageal pH probe is the ‘gold standard’ for diagnosis of GER
esophageal manometry can provide objective data about esophageal motility, which will help
in choosing the appropriate antireflux procedure
Management
Conservative Measures
thickening of formula with cereal
reducing volumes of feedings
head elevation
acid suppression medicines
Surgery
indications include failure to thrive, near-miss SIDS, failure of medical management
in neurologically impaired infants who require a gastrostomy tube for feeding, a
fundoplication is no longer considered routine
a 360-degree wrap (Nissen) is the most effective procedure for controlling GER, but it
also associated with the most complications (gas bloat, dysphagia)
a 270-degree wrap is indicated in infants with poor esophageal motility
laparoscopy is the standard approach for antireflux surgery