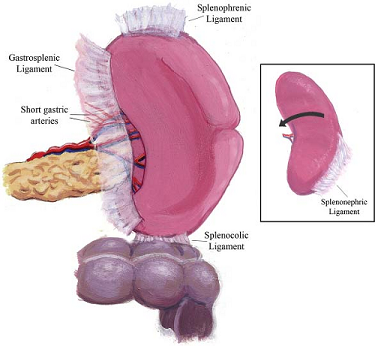
splenophrenic, splenorenal, splenocolic, and gastrosplenic ligaments are the major suspensory
ligaments of the spleen
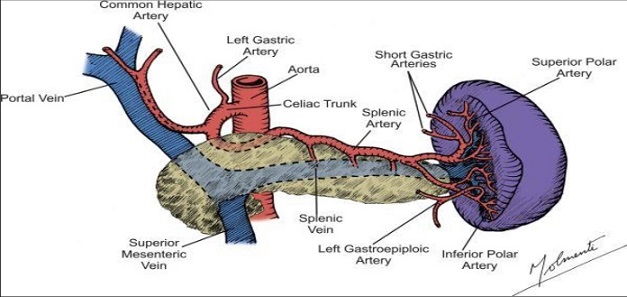
gastrosplenic ligament contains the short gastric and left gastroepiploic vessels; the other
ligaments are relatively avascular, except in portal hypertension
Blood Supply
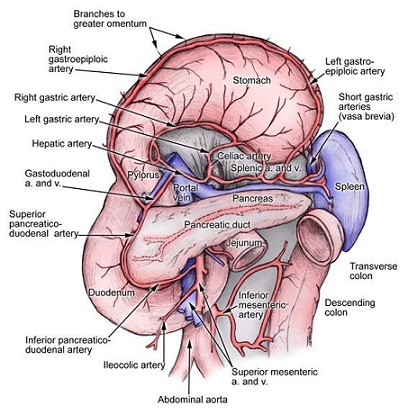
arterial supply is by the splenic artery and the short gastric arteries
venous drainage is via the splenic vein, which joins the superior mesenteric vein to form the portal vein
Internal Architecture
Red Pulp
consists of thin-wall blood vessels called splenic sinuses and a reticular connective
tissue meshwork called splenic cords
within the cords are packed erythrocytes, granulocytes, platelets, macrophages, and plasma cells
White Pulp
lies within the red pulp
zones of lymphatic tissue consisting of lymphocytes, plasma cells, and macrophages
may contain germinal centers
Marginal Zone
interface between the red pulp and white pulp
contains the marginal sinus, which filters material from the white pulp
Microcirculation of the Spleen
splenic artery → trabecular arteries → enter the white pulp as central arteries
central arteries give off numerous branches, some of which terminate in the white pulp,
while others terminate in the marginal zone or red pulp
within the red pulp, blood is collected in the splenic sinuses → trabecular veins →
splenic vein
Open Circulation
arterioles empty blood directly into the pulp cords
blood circulates through the splenic cords before passing through pores in the splenic
sinuses to enter the venous circulation
sluggish nature of the splenic microcirculation facilitates its blood cell management
and immune functions
90% of splenic blood flow is through the open circulation
Closed Circulation
splenic blood follows a continuous endothelial pathway to flow directly into the
splenic sinusoids
10% of splenic circulation
Physiology
Hematopoiesis
produces RBCs and WBCs during early fetal development
process stops after the 5th to 8th month of gestation
hematopoietic potential is retained throughout life
Blood Cell Repair, Removal, and Storage
Repair
RBCs with surface membrane defects such as pits and spurs are repaired in the spleen
nuclear remnants (Howell-Jolly bodies), denatured hemoglobin (Heinz bodies),
and iron granules (Pappenheimer bodies) are removed from circulating RBCs
intracellular parasites such as malaria may be pitted from the RBC by the spleen
Removal
aged RBCs (~ 120 days) that have lost membrane permeability are trapped and destroyed
in the spleen
morphologically abnormal RBCs (spherocytes, sickle cells) are destroyed
blood cells coated with IgG or IgM are destroyed by splenic monocytes
Storage
1/3 of the total platelet pool is normally sequestered in the spleen
this may increase to 80% with splenomegaly
Immune Functions
major site of phagocytosis of antibody coated bacteria or foreign cells
initial site of synthesis of IgM if pre-existing antibodies are lacking
production of specific antibody
major site of clearance of poorly opsonized organisms (encapsulated bacteria)
produces the opsonins properdin and tuftsin
probably plays a major role in removing malignant cells from the circulation
Platelet Disorders
Immune Thrombocytopenic Purpura (ITP)
acquired disorder caused by a circulating antiplatelet antibody that causes destruction of platelets
by the reticuloendothelial system
bone marrow production cannot match destruction to compensate sufficiently
Clinical Presentation
in adults, females > males (3:1); most affected women are < 40 years old
in children, both genders are affected equally
increased incidence in patients with HIV infection
common symptoms include easy bruising, petechiae, gingival bleeding, and nose bleeds
intracranial hemorrhage is a rare but lethal complication
risk of hemorrhage is proportional to the platelet count, and spontaneous bleeding becomes
more common with a platelet count < 20,000
spleen is usually normal-sized
Diagnosis
average platelet count is 33,000; RBC and WBC counts are normal
bone marrow examination shows an increased megakaryocyte count and normal granulocytic and
erythrocytic elements
ITP is a diagnosis of exclusion - must rule out all other causes of thrombocytopenia:
the antiplatelet factor is an IgG antibody directed against a platelet-associated antigen
initial antibody production probably occurs in the spleen
as the immune response becomes more generalized, the bone marrow becomes a major site of
antibody production and is the source of antibody after splenectomy
liver and lymph nodes produce little if any antiplatelet antibody
Platelet Destruction
spleen is the most active site of platelet destruction and is ideally suited for this purpose
for the following reasons:
30% of the total platelet mass resides in the spleen
sluggish splenic microcirculation provides sufficient time for antibody binding
and phagocytosis
since the liver has no resident platelet pool and a rapid microcirculation, it plays a
major role in platelet destruction only when high antibody titers result in heavily sensitized platelets
some platelet destruction, as well as inhibition of thrombopoiesis, may also occur in the bone marrow
Emergency Management of Bleeding
platelets should be administered to control life-threatening bleeding
high-dose gamma globulin is useful but requires several days for a platelet increase to occur
most patients respond to high-dose steroids, but the response may take several days
emergency splenectomy is necessary in patients with CNS bleeding
Chronic ITP in Adults
asymptomatic patients with platelet counts > 50,000 do not require intervention
asymptomatic patients with platelet counts between 30,000 and 50,000 need close
monitoring because they are at risk for progressing to severe thrombocytopenia
treatment is usually initiated when the platelet count < 30,000
Initial Management
initial treatment is prednisone or dexamethasone
two-thirds of patients will respond within 2 – 5 days
only ~ 20% of patients have a complete and sustained remission after the steroids are tapered
Rescue Strategies
required for patients with persistent platelet counts < 20, 000 following steroid treatment
Splenectomy
provides the highest chance of a complete and durable remission, but the failure rate at
5 years is 28%
patients most likely to respond to splenectomy are patients under age 60 and those
patients who have had a good initial response to steroids
Rituximab
monoclonal antibody against B cells (CD20 protein)
used in patients who are not candidates for splenectomy or who chose against it
has a lower rate of sustained remissions than splenectomy
major complication is immunosuppression
may cause reactivation of hepatitis B infection
Thrombopoietin Receptor Agonists
stimulate platelet production in the bone marrow
most patients will have an increased platelet count, but these agents do not induce remission
prolonged maintenance therapy is required
Childhood ITP
often appears after a viral respiratory tract infection
spontaneous and complete remission occurs in most patients
elective splenectomy is indicated if remission has not occurred after 1 year
Perioperative Preparation
Gamma Globulin (IVIG)
given to patients who do not respond to steroids
will significantly raise the platelet count within several days, providing a
therapeutic window for splenectomy
works by saturating the macrophage Fc receptors, producing a transient blockade
of the reticuloendothelial system
Vaccinations
vaccination against common encapsulated organisms may prevent overwhelming postsplenectomy sepsis
all patients should be vaccinated against S. pneumonia, H. influenza, and N. meningitidis 2 weeks
before elective surgery, or within 30 days of an emergent splenectomy
Thromboembolic Precautions
splenic and portal vein thrombosis is a risk after splenectomy
Surgical Considerations
laparoscopic splenectomy is the preferred approach for most elective splenectomies
a nasogastric tube decompresses the stomach and facilitates handling of the short gastric vessels
early ligation of the splenic artery in the lesser sac should be performed before platelets are transfused
must avoid injury to the tail of the pancreas
must search for accessory spleens, which are most commonly located in the splenic hilum, near the splenic vessels
and tail of the pancreas, omentum, and gastrosplenic and gastrocolic ligaments
drains are not used unless there is concern for pancreatic injury
Thrombotic Thrombocytopenic Purpura (TTP)
Pathogenesis
deficiency of the metalloproteinase that cleaves the large multimers of von Willebrand’s factor
results from antimetalloproteinase antibody production
results in platelet clumping and subsequent thrombosis in the microvasculature
plasma exchange results in 70% to 80% remission rates
splenectomy is reserved for the patients who do not respond to plasma exchange, or who relapse
patients who undergo splenectomy have an 8% to 17% relapse rate
Red Blood Cell Disorders
Hereditary Spherocytosis
Pathogenesis
transmitted as an autosomal dominant trait
results in deficiency or dysfunction of spectrin, an RBC cytoskeleton protein
RBCS lose their biconcave shape, become rigid, and lack deformability
more susceptible to trapping and destruction in the spleen
Clinical Manifestations
presents as a hemolytic anemia of varying severity
other signs and symptoms include jaundice and splenomegaly
diagnosis is made by examining the peripheral blood smear, increased reticulocyte count,
and negative Coombs test
Treatment
splenectomy cures the anemia, but not the altered RBC morphology
children should have splenectomy delayed until they are 5 years old to preserve the
immunologic function of the spleen
if gallstones are present, cholecystectomy should be performed at the same time as
splenectomy
hereditary elliptocytosis is another RBC membrane defect abnormality that will often
require splenectomy
Hemoglobinopathies
Sickle Cell Anemia
occurs in patients (blacks) who are homozygous for the HbS gene
under conditions of low oxygen tension, RBCs containing HbS assume a sickle shape
sickled RBCs cause increased blood viscosity and circulatory stasis → thrombosis,
ischemia, necrosis, and organ fibrosis
diagnosis is made by peripheral blood smear and hemoglobin electrophoresis
splenectomy does not affect the sickling process and is reserved for patients with
splenomegaly, excessive splenic sequestration of RBCs, massive infarction,
splenic abscess
if gallstones are present, cholecystectomy should be performed at the same time
as splenectomy
Thalassemia
group of hereditary syndromes in which one of the hemoglobin chains is synthesized
at a markedly reduced rate
beta-thalassemia is the most common type
homozygous patients (thalassemia major) have severe anemia; heterozygous patients
(thalassemia minor) have mild anemia
splenectomy does not alter the basic disease process and is reserved for patients with
symptomatic splenomegaly and pain from splenic infarcts
incidence of overwhelming postsplenectomy sepsis is high in this population
Autoimmune Hemolytic Anemia
Pathogenesis
results from autoantibodies to RBC antigens
usually seen in association with other autoimmune diseases such as lupus
may also be associated with drug exposures
Clinical manifestations
anemia, fluctuating jaundice, and splenomegaly
Diagnosis
patients have an elevated reticulocyte count
peripheral smear shows many spherocytes
direct Coombs test is positive, which indicates the presence of antibody on
the red cell surface
anti-red cell antibodies may be classified as ‘warm’ reactive or ‘cold’ reactive
Treatment
steroid therapy is often effective
splenectomy should be considered if steroids are ineffective or contraindicated
patients with ‘cold’ antibodies do not respond to splenectomy
after splenectomy, 20% of patients will have long-term remissions, and 50% will have a
significant decrease in steroid requirement
Lymphomas and Leukemias
Indications for Splenectomy
massive splenomegaly may cause abdominal pain, early satiety, abdominal distention, or atraumatic splenic rupture
hypersplenism, which is defined as splenomegaly with associated cytopenias
Massive Splenomegaly
Late Complications of Splenectomy
Thrombocytosis
occurs particularly in patients with myeloproliferative disorders
can result in thrombosis of the splenic, portal, and renal veins
may be a significant risk for DVT and pulmonary embolism
Overwhelming Postsplenectomy Sepsis (OPSS)
most common fatal late complication of splenectomy
may occur at any time after splenectomy
most commonly involved pathogen is S. pneumonia (50% to 90% of cases)
other isolated organisms include H. influenzae, N. meningitidis, Streptococcus, Salmonella
Clinical Manifestations
fever, rigors, chills
nonspecific symptoms include sore throat, malaise, myalgias, vomiting
pneumonia and meningitis may be present
often there is no identifiable site of infection
progression to full-blown sepsis is rapid, with a 50% to 70% mortality rate
Additional Risk Factors for OPSS
splenectomy for malignant disease or hematologic conditions results in more OPSS than
splenectomy for trauma
children < 4 are at great risk
Immunization
for patients between 2 and 64 years old, the recommended vaccines are the
23-valent pneumococcal, meningococcal, and Haemophilus vaccines
timing should be at least 2 weeks before elective surgery, or within 30 days postoperatively
in emergency cases
routine revaccination of immunocompetent patients is not recommended by the CDC
selected high-risk patients are recommended to have one PPV23 revaccination dose after
5 years
Antibiotic Prophylaxis
in children, routine prophylaxis with penicillin for at least 2 years after splenectomy
is common practice
some recommend life-long prophylaxis in both adults and children
another recommendation is to provide adults with a supply of oral antibiotics to take if
they develop a febrile illness
References
Sabiston, 20th ed., pgs 1556 - 1568
Schwartz, 10th ed., pgs 1423 - 1445
Cameron, 13th ed., pgs 605 – 612
UpToDate. Immune Thrombocytopenia (ITP) in Adults: Initial Treatment and Prognosis.
Donald M. Arnold, MD, MSc. Sep 20, 2019. Pgs 1 – 25
UpToDate. Immune Thrombocytopenia (ITP) in Adults: Second-Line and Subsequent therapies.
James N. George, MD, Donald M. Arnold, MD, MSc. May 04, 2020. Pgs 1 – 42