Molecular Biology of Cancer
- Cellular Homeostasis
- to achieve homeostasis in tissues, renewable cell populations must perform 4 related functions:
- must proliferate with proper timing and fidelity of DNA repair
- must differentiate in a pattern consistent with normal function of the tissue
- proliferation and involution rates must be balanced
- must repair any damage resulting from exposure to mutagens such as radiation, chemicals, or viruses
- a defect in any of these functions can result in tumor formation
- Cancer Phenotype
- tumors are clonal in origin
- loss of contact inhibition
- greater autonomy from growth factors
- nuclear and cellular polymorphism
- loss of cellular polarity
- variation in DNA content from cell to cell (aneuploidy)
- development of invasive properties
- neovascularization (angiogenesis)
- resistance to apoptosis
- cell cycle differences
- Genetic Alterations
- Multistep Hypothesis
- cancer is fundamentally an alteration in genes that control cellular function
- development of the malignant phenotype is multifactorial
- some abnormal genes can be inherited as germline defects (cancer susceptibility genes)
- other genetic changes (somatic mutations) are acquired through interaction with agents
that alter the cellular genome (radiation, mutagenic chemicals, viruses)
- carcinogenesis requires the successive accumulation of genetic defects
(multi-hit hypothesis) that result in the altered cellular growth and differentiation
characteristic of the malignant phenotype
- each mutation confers a growth advantage to the cell and enhances the rate at which subsequent
mutations occur
- the genetic changes necessary to develop cancer are best understood for colon cancer and melanoma
- Adenoma-Carcinoma Sequence in Colorectal Cancer
- most colorectal cancers arise from adenomatous polyps that become dysplastic and then malignant (invasive)
- mutations in the adenomatous polyposis coli (APC) gene occur early, kras and DCC gene mutations are
intermediate steps, and p53 gene mutations occur late

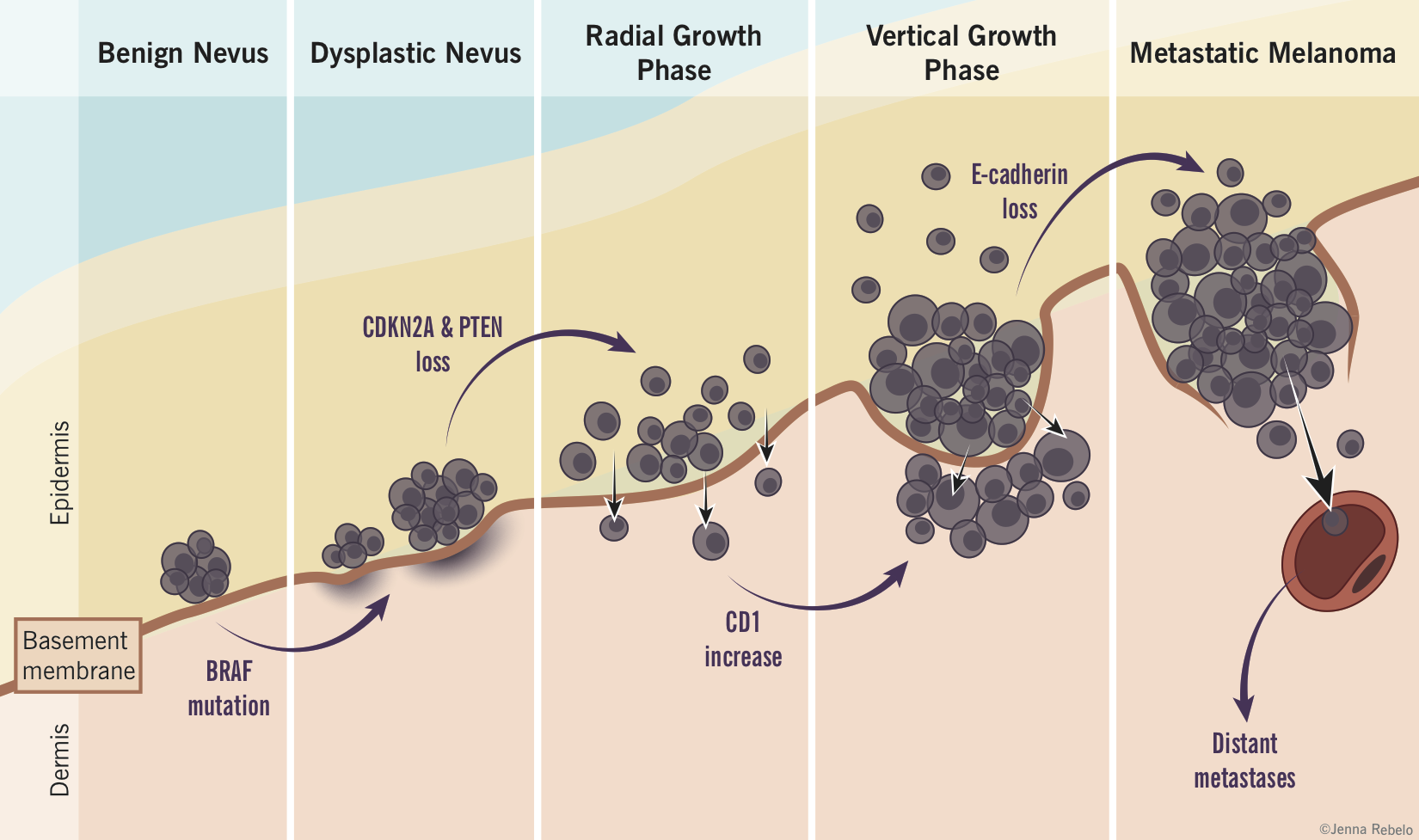
- Nevus-Melanoma Sequence
- nevi without atypia may progress to dysplastic nevi
- some dysplastic nevi may progress to the radial growth phase of melanoma
- some superficial melanomas may develop a clone of fast growing cells that expand in the vertical direction
- some of these melanomas may then develop the ability to metastasize
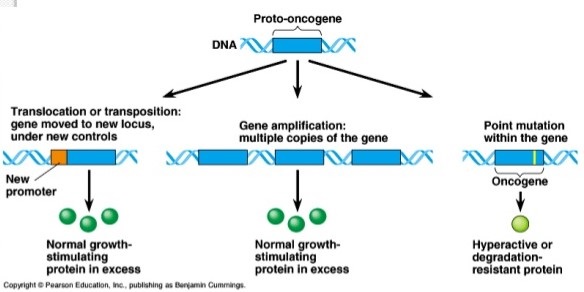
- Oncogenes
- genes that promote the transformation of normal cells into tumor cells
- oncogenes are homologs of normal cellular genes that participate in cell growth and cell cycle
regulation
- normal host genes are termed protooncogenes
- a mutational change in an oncogene leads to constitutive activation of the gene, which then
results in uncontrolled cellular proliferation
- oncogenes are gain in function mutations
- Growth Factors
- transformation of normal cells to malignant
cells may result from increased production of stimulatory growth factors or
decreased production of inhibitory growth factors (TGF-β)
- growth factors function by binding to a growth factor receptor
- activation of the growth factor receptor leads to activation of the intrinsic receptor enzymatic activity and signal transduction follows
- sis oncogene displays significant homology to the β-chain of PDGF
- overproduction of epidermal growth factor (EGF) will result in cellular transformation
- Growth Factor Receptor Tyrosine Kinases
- prototypical example is the EGFR group (HER1 through HER4)
- constitutive activation of a growth factor receptor can also lead to tumor formation
- mutation of the erb protooncogene results in a defective EGFR that can be activated without EGF, leading to activation
of tyrosine kinase and downstream signaling activation
- HER2 gene is amplified in 30% of breast and ovarian cancers and is associated with a poorer prognosis
- HER2 is a target for therapy by a recombinant antibody called trastuzumab (Herceptin)
- Ras GTPases
- G proteins function as mediators between G-protein-linked receptors and membrane-associated enzymes
- after ligand binding, the G protein releases GDP and binds GTP
- G protein then dissociates and activates specific enzymes (cAMP, phospholipase C, protein kinase C)
- ras oncogenes have significant homology to G proteins
- because of a mutation at the GTP binding site, the ras protein does not hydrolyze GTP and so is continuously activated
- ras oncogenes are the second most commonly expressed oncogenes in human malignancies
- Nuclear Transcription Factors
- directly alter gene expression by binding to DNA to effect gene transcription
- mutated oncogenes may upregulate or act as negative inhibitors by blocking the genes necessary for normal growth
and differentiation
- c-myc is a direct regulator of the cell cycle and stimulates the cell to progress from G1 into S phase
- Tumor Suppressor Genes and Cancer Predisposition Syndromes
- normal effect is to keep cellular growth in check
- loss of function leads to tumor formation
- most of the inherited cancer predisposition syndromes involve inheritance of one mutant allele and one
normal allele of a tumor suppressor gene
- additionally, expression of the malignant phenotype requires loss of the normal allele through somatic mutation
(‘two-hit’ hypothesis)
- in keeping with the multistep hypothesis, additional oncogenic mutations are required to develop the malignant phenotype
(ras, c-myc, etc)
- Hereditary Retinoblastoma and the Rb Gene
- usually presents in the first year of life
- Rb is a nuclear protein that regulates the entry of cells into S-phase
- Rb inhibits the ability of E2F transcription factor to bind DNA and initiate DNA synthesis
- in late G1, Rb is phosphorylated by CDKs and releases E2F
- E2F is a transcription factor responsible for leading the cell from G1 into S phase
- a mutation in Rb leads to unregulated cell growth through increased E2F activity
- p53
- most frequently mutated gene in human cancer
- p53 is a DNA-binding protein responsible for protecting the integrity of the cell’s genome
- p53 is able to activate genes that halt the cell cycle in response to damage, allowing adequate
time for DNA repair prior to replication
- in cases of severe damage, p53 is capable of triggering apoptosis, eliminating damaged cells before
replication can take place
- intact p53 function is crucial for tumor prevention
- inactivation of p53 is one of the most detectable genetic defects in sporadic tumors
- inherited mutations of p53 (Li-Fraumeni syndrome) result in tumors of the breast, brain, adrenal,
sarcoma, and leukemia
- Familial Adenomatous Polyposis and the APC Gene
- autosomal dominant
- characterized by the development of hundreds of colon polyps and the nearly 100
percent progression to colorectal cancer in untreated individuals
- phenotype may also include duodenal adenomas and carcinomas, desmoid tumors, mandibular osteomas
- results from a germline mutation in the APC gene
- mutations in the APC gene have been found in all types of colon polyps and in over 80% of sporadic colon cancers
- APC regulates β-catenin, which is a nuclear transcription factor
- loss of APC function results in nuclear accumulation of β-catenin, leading to cellular proliferation
- Familial Breast and Ovarian Cancer
- cancer susceptibility genes include BRCA-1 and BRCA-2
- lifetime risk of breast cancer is 80%
- BRCA-1 has a 60% risk of ovarian cancer; BRCA-2 has a 27% risk
- genes are involved in cell cycle control, DNA damage repair
- Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome)
- accounts for 2% of all colon cancers
- autosomal dominant with high penetrance
- caused by mutations in DNA mismatch repair genes
- cancers have a right-sided predominance that appear at an earlier age (median age of 45 years)
- extracolonic malignancies occur, especially of the ovary and endometrium
- Regulation of Cell Death
- cell proliferation and involution rates must be balanced
- defects resulting in loss of normal apoptosis are associated with tumor formation
- induction of apoptosis may result from activation of death receptor (Fas) pathways or death receptor
independent pathways (hypoxia, DNA damage, etc)
- apoptotic pathways involve the release of cytochrome c from mitochondria, which then activates various
caspases (proteases)
- most tumors have defects in cell-death signaling pathways and are resistant to cell death
- bcl-2 oncogene is a major repressor of cell death
- p53 may induce growth arrest or apoptosis depending upon the cellular circumstances
- Immortality
- except for stem cells, germline cells, and activated lymphocytes, normal cells have a limited replicative potential
- for a population of transformed cells to develop into a macroscopic tumor, the program that limits cell doublings
must be turned off
- the number of doublings is controlled by telomeres, which prevent end to end chromosomal fusion
- each DNA replication results in telomeric shortening
- eventually, the progressive shortening of telomeres causes them to lose their ability to prevent chromosomal fusion
- chromosomal fusion inevitably leads to cell death
- telomerase, normally inactive in most cells, elongates telomeric DNA
- many tumors have an elevated telomerase activity
- if the telomeres are maintained above a certain critical level, the cell will have unlimited replicative potential
and be immortal
- Angiogenesis
- angiogenesis is a highly regulated process
- endogenous angiogenesis inhibitor factors prevent tumors from expanding beyond microscopic size
- angiogenic inhibitors counteract angiogenic signals from the tumor cells
- angiogenic activity is induced by growth factors such as VEGF, FGF, PDGF
- at least 26 different angiogenic inhibitors have been identified
- tumor growth occurs when angiogenesis stimulators overwhelm the angiogenesis inhibitors
- Avastin (bevacizumab), an anti-VEGF antibody, prolongs survival is patients with advanced colon cancer
- Tissue Invasion and Metastasis
- distant metastasis is the cause of 90% of cancer deaths
- entire process is not well understood, but many steps must occur:
- tumor cells must detach from the primary tumor and infiltrate into the bloodstream or
lymphatics (intravasation)
- tumor cells must survive immune destruction as well as other cellular defenses
- after arriving in the recipient organ capillary bed, the cells must extravasate into the organ parenchyma
- tumor cells must then develop a blood supply and evade host defenses
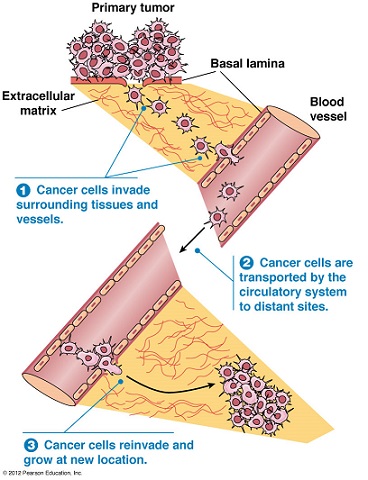
- Tissue Invasion
- characterized by changes in the extracellular matrix (ECM) and its interaction with tumor cells
- cell-cell and cell-matrix interactions are mediated through cell adhesion molecules (CAMs)
- E-cadherin is responsible for cell polarity and organization of epithelium
- E-cadherin activity is lost in most epithelial tumors, and may be a prerequisite for tumor invasion and
metastasis
- activation of extracellular proteases (collagenases, MMPs) are also critical to invasiveness and metastatic
potential
- Metastasis
- metastasis is not a random event: certain tumor cells (seed) have a propensity for a certain organ (soil)
- tumor cells may only grow in environments where favorable growth factors exist
- another theory proposes that endothelial cells in certain organs express adhesion molecules that bind tumor
cells and permit extravasation
- chemokines expressed by target organs may selectively attract tumor cells that express receptors for the chemokines
- Evasion of Immune Destruction
- much experimental and observational evidence demonstrates that the immune system can recognize and eliminate primary
tumors
- however, the immune system fails to eliminate tumor cells with reduced immunogenicity, thereby selecting for tumor
variants that have acquired immune evasion mechanisms
- Elimination
- innate immune system is the first line of defense against transformed cells (macrophages, NK cells)
- activated innate immune cells produce IFN-γ, which has direct antitumor effects
- tumor lysis makes antigen available to trigger an adaptive immune response by T and B lymphocytes
- Equilibrium
- innate and adaptive immune systems do not always completely eliminate the tumor
- some tumor cells in the tumor mass may have reduced immunogenicity and escape destruction
- equilibrium phase is characterized by a balance between tumor growth and tumor elimination
- Escape
- over time, tumor variants may be selected that are able to escape immune detection and destruction
- host-related factors include treatment related immunosuppression, acquired or inherited immunodeficiency,
and aging
- tumor-related escape mechanisms include loss of MHC alleles, reduced antigen processing and/or presentation,
secretion of immunosuppressive factors (TGF-β, IL-10), stimulation of suppressor cells
Carcinogenesis
- Chemical Agents
- most chemical carcinogens require metabolic activation for their carcinogenic effects
- DNA is the primary target of chemical carcinogens
- ability of a compound to cause mutations is termed mutagenic potential
- agents called promoters may augment the carcinogenicity of some chemicals
- some proven chemical carcinogens include aflatoxins, asbestos, benzene, tobacco smoke
- some proven pharmaceutical carcinogens include tamoxifen, estrogens, azathioprine
- Radiation
- UV Radiation
- known risk factor for skin cancers: BCCs, SCCAs, possibly melanoma
- UVB is the most important wavelength for carcinogenicity
- other risk factors include length of exposure and amount of melanin present
- UVB causes DNA damage that must be repaired in a multistep process
- it is postulated that excessive sun exposure causes the DNA repair system to be
overwhelmed, with the result that some damaged DNA goes unrepaired
- Ionizing Radiation (IR)
- includes electromagnetic (x-rays, gamma rays) and particulate (alpha particles, beta particles, protons, neutrons)
forms
- IR is a carcinogen and therapeutic agent – at low doses, it is a carcinogen; at high doses, it can stop tumor growth
- IR leads to persistent activation of the microenvironment, leading to long-term production of reactive oxygen or
nitrogen species by tissue macrophages or neutrophils
- long-term exposure to these inflammatory products may lead to chromosomal abnormalities or gene mutations
- different tissues have different vulnerabilities to radiation-induced carcinogenesis – bone marrow (leukemias)
and the thyroid gland have the highest
- Infectious Carcinogens
- cause cancer via different mechanisms: direct transformation, expression of oncogenes that interfere with cell cycle checkpoints
or DNA repair, expression of growth factors, alteration of the immune system
- Viral Carcinogenesis
- 15% of tumors worldwide are caused by viruses
- cervical cancer (HPV) and hepatocellular cancer (HBV, HCV) make up most these tumors
- tumor viruses establish persistent infections in natural hosts
- long latent periods elapse between infection and tumor appearance
- direct-acting viruses contain one or more oncogenes that modulate growth control pathways in cells
- indirect-acting viruses don’t appear to contain an oncogene
- viruses are rarely complete carcinogens
- host factors are important determinants of virus-induced tumorigenesis
- Bacterial Carcinogenesis
- H. pylori infection is the most important risk factor for gastric cancer
- chronic inflammatory response is an important mechanism whereby infection can lead to neoplasia
- gastric microenvironment – hypoacidity – is also important
- Chronic Inflammation
- chronic inflammation in the absence of infection has long been linked to development of cancer
- examples include development of SCCA of the skin in areas of chronic ulceration (Marjolin’s ulcer) and
colon cancer in ulcerative colitis patients
- release of proinflammatory agents such as cytokines, prostaglandins, and interleukins may indirectly
promote survival of transformed cells
Tumor Markers
- Overview
- indicators of cellular, biochemical, molecular, or genetic alterations of cancer
- surrogate measures of the biology of the cancer
- the ideal tumor marker would have the following characteristics:
- be expressed exclusively by the particular tumor
- be inexpensive and easy to collect
- be diagnostic, distinguishing benign from malignant disease
- correlate with the amount of tumor present
- be prognostic and allow more accurate staging
- guide choice of therapy
- predict response to therapy
- Protein Tumor Markers
- clinical use limited by poor sensitivity and specificity
- serum levels generally correlate with tumor burden because they are shed from the expanding neoplasm
- most are not useful for screening because of low sensitivity in early-stage disease
- preoperative levels have prognostic value since they correlate with tumor burden
- most common application is monitoring patients for recurrent disease
- also useful in monitoring response to chemotherapy – patients whose CEA levels fall during
chemotherapy survive longer than those whose CEA levels do not change or rise
- current tumor markers in clinical use include CEA (colorectal), AFP (hepatocellular), CA 19-9 (pancreatic), CA-125 (ovarian),
PSA (prostate)
- DNA-Based Markers
- specific mutations in oncogenes, tumor suppressor genes, and mismatch repair genes can serve as biomarkers
- germline mutations can be screened for and preventative surgery offered (RET in MEN2, APC in FAP, BRCA1 and BRCA2)
- estrogen and progesterone receptors guide aromatase inhibitor therapy in breast cancer
- somatic mutations can be screened for and are starting to profoundly change cancer treatment
- c-kit predicts response to imatinib (Sulindac) in GIST tumors
- HER-2/neu amplification guides treatment with Herceptin in breast cancer
- KRAS gene predicts lack of response to anti-EGFR monoclonal antibody therapy in patients with metastatic
colorectal cancer
References
- Sabiston, 20th ed., pgs 677 - 702
- O’Leary, 4th ed., pgs 188 – 207
- The Molecular Basis of Cancer-Cell Behavior. www.ncbi.nim.nih.gov/books/NBK26902/
- Molecular genetics of colorectal cancer. Frucht, Harold MD, Lucas, Aimee MD. UpToDate. Jan 15, 2019. Pgs 1-37.